
α-Amylase (Porcine Pancreatic)



12 g / 900 KU
Prices exclude VAT
Available for shipping
| Content: | 12 g / 900 KU or 4 g / 300 KU |
| Shipping Temperature: | Ambient |
| Storage Temperature: | Below -10oC |
| Formulation: | In powder form |
| Physical Form: | Powder |
| Stability: | > 1 year under recommended storage conditions |
| Enzyme Activity: | α-Amylase |
| EC Number: | 3.2.1.1 |
| CAZy Family: | GH13 |
| CAS Number: | 9000-90-2 |
| Synonyms: | alpha-amylase; 4-alpha-D-glucan glucanohydrolase |
| Source: | Porcine Pancreatic |
| Molecular Weight: | 54,000 |
| Expression: | Purified from Porcine pancreas |
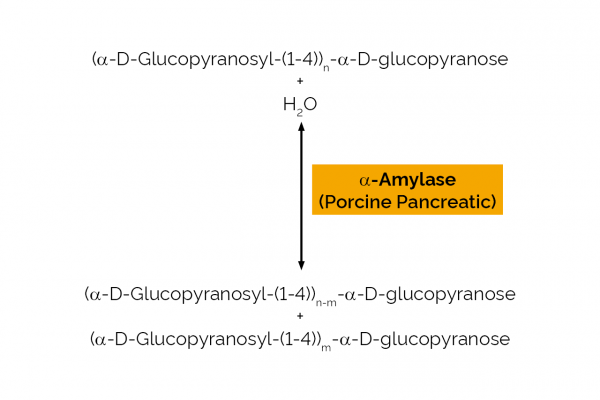
| Specificity: | endo-hydrolysis of α-1,4-D-glucosidic linkages in starch. |
| Specific Activity: | ca. 75,000 U/g (40oC, pH 6.9 on Ceralpha reagent) |
| Unit Definition: | One Unit of α-amylase activity is the amount of enzyme required to release one µmole of p-nitrophenol from blocked p-nitrophenyl-maltoheptaoside per minute, in the presence of excess α-glucosidase at pH 6.9 and 40oC. |
| Temperature Optima: | 50oC |
| pH Optima: | 6.9 |
| Application examples: | For use in the Megazyme Integrated Total Dietary Fiber kit. |
The E-PANAA-4G pack size has been discontinued (read more).
High purity α-Amylase (Porcine Pancreatic) for use in research, biochemical enzyme assays and in vitro diagnostic analysis.
For use in the Megazyme Integrated Dietary Fiber assay.
Find out more available CAZyme products.
Measurement of available carbohydrates, digestible, and resistant starch in food ingredients and products.
McCleary, B. V., McLoughlin, C., Charmier, L. M. J. & McGeough, P. (2019). Cereal Chemistry, 97(1), 114-137.
Background and objectives: The importance of selectively measuring available and unavailable carbohydrates in the human diet has been recognized for over 100 years. The levels of available carbohydrates in diets can be directly linked to major diseases of the Western world, namely Type II diabetes and obesity. Methodology for measurement of total carbohydrates by difference was introduced in the 1880s, and this forms the basis of carbohydrate determination in the United States. In the United Kingdom, a method to directly measure available carbohydrates was introduced in the 1920s to assist diabetic patients with food selection. The aim of the current work was to develop simple, specific, and reliable methods for available carbohydrates and digestible starch (and resistant starch). The major component of available carbohydrates in most foods is digestible starch. Findings: Simple methods for the measurement of rapidly digested starch, slowly digested starch, total digestible starch, resistant starch, and available carbohydrates have been developed, and the digestibility of phosphate cross‐linked starch has been studied in detail. The resistant starch procedure developed is an update of current procedures and incorporates incubation conditions with pancreatic α‐amylase (PAA) and amyloglucosidase (AMG) that parallel those used AOAC Method 2017.16 for total dietary fiber. Available carbohydrates are measured as glucose, fructose, and galactose, following complete and selective hydrolysis of digestible starch, maltodextrins, maltose, sucrose, and lactose to glucose, fructose, and galactose. Sucrose is hydrolyzed with a specific sucrase enzyme that has no action on fructo‐oligosaccharides (FOS). Conclusions: The currently described “available carbohydrates” method together with the total dietary fiber method (AOAC Method 2017.16) allows the measurement of all carbohydrates in food products, including digestible starch. Significance and novelty: This paper describes a simple and specific method for measurement of available carbohydrates in cereal, food, and feed products. This is the first method that provides the correct measurement of digestible starch and sucrose in the presence of FOS. Such methodology is essential for accurate labeling of food products, allowing consumers to make informed decisions in food selection.
Hide AbstractModification to AOAC Official Methods 2009.01 and 2011.25 to allow for minor overestimation of low molecular weight soluble dietary fiber in samples containing starch.
McCleary, B. V. (2014). Journal of AOAC International, 97(3), 896-901.
AOAC Official Methods 2009.01 and 2011.25 have been modified to allow removal of resistant maltodextrins produced on hydrolysis of various starches by the combination of pancreatic α-amylase and amyloglucosidase (AMG) used in these assay procedures. The major resistant maltodextrin, 63,65-di-α-D-glucosyl maltopentaose, is highly resistant to hydrolysis by microbial α-glucosidases, isoamylase, pullulanase, pancreatic, bacterial and fungal α-amylase and AMG. However, this oligosaccharide is hydrolyzed by the mucosal α-glucosidase complex of the pig small intestine (which is similar to the human small intestine), and thus must be removed in the analytical procedure. Hydrolysis of these oligosaccharides has been by incubation with a high concentration of a purified AMG at 60°C. This incubation results in no hydrolysis or loss of other resistant oligosaccharides such as FOS, GOS, XOS, resistant maltodextrins (e.g., Fibersol 2) or polydextrose. The effect of this additional incubation with AMG on the measured level of low molecular weight soluble dietary fiber (SDFS) and of total dietary fiber in a broad range of samples is reported. Results from this study demonstrate that the proposed modification can be used with confidence in the measurement of dietary fiber.
Hide AbstractMeasurement of total dietary fiber using AOAC method 2009.01 (AACC International approved method 32-45.01): Evaluation and updates.
McCleary, B. V., Sloane, N., Draga, A. & Lazewska, I. (2013). Cereal Chemistry, 90(4), 396-414.
The Codex Committee on Methods of Analysis and Sampling recently recommended 14 methods for measurement of dietary fiber, eight of these being type I methods. Of these type I methods, AACC International Approved Method 32-45.01 (AOAC method 2009.01) is the only procedure that measures all of the dietary fiber components as defined by Codex Alimentarius. Other methods such as the Prosky method (AACCI Approved Method 32-05.01) give similar analytical data for the high-molecular-weight dietary fiber contents of food and vegetable products low in resistant starch. In the current work, AACCI Approved Method 32-45.01 has been modified to allow accurate measurement of samples high in particular fructooligosaccharides: for example, fructotriose, which, in the HPLC system used, chromatographs at the same point as disaccharides, meaning that it is currently not included in the measurement. Incubation of the resistant oligosaccharides fraction with sucrase/β-galactosidase removes disaccharides that interfere with the quantitation of this fraction. The dietary fiber value for resistant starch type 4 (RS4), varies significantly with different analytical methods, with much lower values being obtained with AACCI Approved Method 32-45.01 than with 32-05.01. This difference results from the greater susceptibility of RS4 to hydrolysis by pancreatic α-amylase than by bacterial α-amylase, and also a greater susceptibility to hydrolysis at lower temperatures. On hydrolysis of samples high in starch in the assay format of AACCI Approved Method 32-45.01 (AOAC method 2009.01), resistant maltodextrins are produced. The major component is a heptasaccharide that is highly resistant to hydrolysis by most of the starch-degrading enzymes studied. However, it is hydrolyzed by the maltase/amyloglucosidase/isomaltase enzyme complex present in the brush border lining of the small intestine. As a consequence, AOAC methods 2009.01 and 2011.25 (AACCI Approved Methods 32-45.01 and 32-50.01, respectively) must be updated to include an additional incubation with amyloglucosidase to remove these oligosaccharides.
Hide AbstractDetermination of insoluble, soluble, and total dietary fiber (codex definition) by enzymatic-gravimetric method and liquid chromatography: Collaborative Study.
McCleary, B. V., DeVries, J. W., Rader, J. I., Cohen, G., Prosky, P., Mugford, D. C., Champ, M. & Okuma, K. (2012). Journal of AOAC International, 95(3), 824-844.
A method for the determination of insoluble (IDF), soluble (SDF), and total dietary fiber (TDF), as defined by the CODEX Alimentarius, was validated in foods. Based upon the principles of AOAC Official MethodsSM 985.29, 991.43, 2001.03, and 2002.02, the method quantitates water-insoluble and water-soluble dietary fiber. This method extends the capabilities of the previously adopted AOAC Official Method 2009.01, Total Dietary Fiber in Foods, Enzymatic-Gravimetric-Liquid Chromatographic Method, applicable to plant material, foods, and food ingredients consistent with CODEX Definition 2009, including naturally occurring, isolated, modified, and synthetic polymers meeting that definition. The method was evaluated through an AOAC/AACC collaborative study. Twenty-two laboratories participated, with 19 laboratories returning valid assay data for 16 test portions (eight blind duplicates) consisting of samples with a range of traditional dietary fiber, resistant starch, and nondigestible oligosaccharides. The dietary fiber content of the eight test pairs ranged from 10.45 to 29.90%. Digestion of samples under the conditions of AOAC 2002.02 followed by the isolation, fractionation, and gravimetric procedures of AOAC 985.29 (and its extensions 991.42 and 993.19) and 991.43 results in quantitation of IDF and soluble dietary fiber that precipitates (SDFP). The filtrate from the quantitation of water-alcohol-insoluble dietary fiber is concentrated, deionized, concentrated again, and analyzed by LC to determine the SDF that remains soluble (SDFS), i.e., all dietary fiber polymers of degree of polymerization = 3 and higher, consisting primarily, but not exclusively, of oligosaccharides. SDF is calculated as the sum of SDFP and SDFS. TDF is calculated as the sum of IDF and SDF. The within-laboratory variability, repeatability SD (Sr), for IDF ranged from 0.13 to 0.71, and the between-laboratory variability, reproducibility SD (sR), for IDF ranged from 0.42 to 2.24. The within-laboratory variability sr for SDF ranged from 0.28 to 1.03, and the between-laboratory variability sR for SDF ranged from 0.85 to 1.66. The within-laboratory variability sr for TDF ranged from 0.47 to 1.41, and the between-laboratory variability sR for TDF ranged from 0.95 to 3.14. This is comparable to other official and approved dietary fiber methods, and the method is recommended for adoption as Official First Action.
Hide AbstractDetermination of total dietary fiber (CODEX definition) by enzymatic-gravimetric method and liquid chromatography: collaborative study.
McCleary, B. V., DeVries, J. W., Rader, J. I., Cohen, G., Prosky, L., Mugford, D. C., Champ, M. & Okuma, K. (2010). Journal of AOAC International, 93(1), 221-233.
A method for the determination of total dietary fiber (TDF), as defined by the CODEX Alimentarius, was validated in foods. Based upon the principles of AOAC Official MethodsSM 985.29, 991.43, 2001.03, and 2002.02, the method quantitates high- and low-molecular-weight dietary fiber (HMWDF and LMWDF, respectively). In 2007, McCleary described a method of extended enzymatic digestion at 37°C to simulate human intestinal digestion followed by gravimetric isolation and quantitation of HMWDF and the use of LC to quantitate low-molecular-weight soluble dietary fiber (LMWSDF). The method thus quantitates the complete range of dietary fiber components from resistant starch (by utilizing the digestion conditions of AOAC Method 2002.02) to digestion resistant oligosaccharides (by incorporating the deionization and LC procedures of AOAC Method 2001.03). The method was evaluated through an AOAC collaborative study. Eighteen laboratories participated with 16 laboratories returning valid assay data for 16 test portions (eight blind duplicates) consisting of samples with a range of traditional dietary fiber, resistant starch, and nondigestible oligosaccharides. The dietary fiber content of the eight test pairs ranged from 11.57 to 47.83. Digestion of samples under the conditions of AOAC Method 2002.02 followed by the isolation and gravimetric procedures of AOAC Methods 985.29 and 991.43 results in quantitation of HMWDF. The filtrate from the quantitation of HMWDF is concentrated, deionized, concentrated again, and analyzed by LC to determine the LMWSDF, i.e., all nondigestible oligosaccharides of degree of polymerization 3. TDF is calculated as the sum of HMWDF and LMWSDF. Repeatability standard deviations (Sr) ranged from 0.41 to 1.43, and reproducibility standard deviations (SR) ranged from 1.18 to 5.44. These results are comparable to other official dietary fiber methods, and the method is recommended for adoption as Official First Action.
Hide AbstractMcCleary, B. V., Mills, C. & Draga, A. (2009). Quality Assurance and Safety of Crops & Foods, 1(4), 213–224.
An integrated total dietary fibre (TDF) method, consistent with the recently accepted CODEX definition of dietary fibre, has been developed. The CODEX Committee on Nutrition and Foods for Special Dietary Uses (CCNFSDU) has been deliberating for the past 8 years on a definition for dietary fibre that correctly reflects the current consensus thinking on what should be included in this definition. As this definition was evolving, it became evident to us that neither of the currently available methods for TDF (AOAC Official Methods 985.29 and 991.43), nor a combination of these and other methods, could meet these requirements. Consequently, we developed an integrated TDF procedure, based on the principals of AOAC Official Methods 2002.02, 991.43 and 2001.03, that is compliant with the new CODEX definition. This procedure quantitates high- and low-molecular weight dietary fibres as defined, giving an accurate estimate of resistant starch and non-digestible oligosaccharides also referred to as low-molecular weight soluble dietary fibre. In this paper, the method is discussed, modifications to the method to improve simplicity and reproducibility are described, and the results of the first rounds of interlaboratory evaluation are reported.
Hide AbstractAn integrated procedure for the measurement of total dietary fibre (including resistant starch), non-digestible oligosaccharides and available carbohydrates.
McCleary, B. V. (2007). Analytical and Bioanalytical Chemistry, 389(1), 291-308.
A method is described for the measurement of dietary fibre, including resistant starch (RS), non-digestible oligosaccharides (NDO) and available carbohydrates. Basically, the sample is incubated with pancreatic α-amylase and amyloglucosidase under conditions very similar to those described in AOAC Official Method 2002.02 (RS). Reaction is terminated and high molecular weight resistant polysaccharides are precipitated from solution with alcohol and recovered by filtration. Recovery of RS (for most RS sources) is in line with published data from ileostomy studies. The aqueous ethanol extract is concentrated, desalted and analysed for NDO by high-performance liquid chromatography by a method similar to that described by Okuma (AOAC Method 2001.03), except that for logistical reasons, D-sorbitol is used as the internal standard in place of glycerol. Available carbohydrates, defined as D-glucose, D-fructose, sucrose, the D-glucose component of lactose, maltodextrins and non-resistant starch, are measured as D-glucose plus D-fructose in the sample after hydrolysis of oligosaccharides with a mixture of sucrase/maltase plus β-galactosidase.
Hide AbstractMeasurement of novel dietary fibres.
McCleary, B. V. & Rossiter, P. (2004). Journal of AOAC International, 87(3), 707-717.
With the recognition that resistant starch (RS) and nondigestible oligosaccharides (NDO) act physiologically as dietary fiber (DF), a need has developed for specific and reliable assay procedures for these components. The ability of AOAC DF methods to accurately measure RS is dependent on the nature of the RS being analyzed. In general, NDO are not measured at all by AOAC DF Methods 985.29 or 991.43, the one exception being the high molecular weight fraction of fructo-oligosaccharides. Values obtained for RS, in general, are not in good agreement with values obtained by in vitro procedures that more closely imitate the in vivo situation in the human digestive tract. Consequently, specific methods for the accurate measurement of RS and NDO have been developed and validated through interlaboratory studies. In this paper, modifications to AOAC fructan Method 999.03 to allow accurate measurement of enzymically produced fructo-oligosaccharides are described. Suggested modifications to AOAC DF methods to ensure complete removal of fructan and RS, and to simplify pH adjustment before amyloglucosidase addition, are also described.
Hide AbstractMcCleary, B. V. (2003). Proceedings of the Nutrition Society, 62, 3-9.
The 'gold standard' method for the measurement of total dietary fibre is that of the Association of Official Analytical Chemists (2000; method 985.29). This procedure has been modified to allow measurement of soluble and insoluble dietary fibre, and buffers employed have been improved. However, the recognition of the fact that non-digestible oligosaccharides and resistant starch also behave physiologically as dietary fibre has necessitated a re-examination of the definition of dietary fibre, and in turn, a re-evaluation of the dietary fibre methods of the Association of Official Analytical Chemists. With this realisation, the American Association of Cereal Chemists appointed a scientific review committee and charged it with the task of reviewing and, if necessary, updating the definition of dietary fibre. It organised various workshops and accepted comments from interested parties worldwide through an interactive website. More recently, the (US) Food and Nutrition Board of the Institute of Health, National Academy of Sciences, under the oversight of the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes, assembled a panel to develop a proposed definition(s) of dietary fibre. Various elements of these definitions were in agreement, but not all. What was clear from both reviews is that there is an immediate need to re-evaluate the methods that are used for dietary fibre measurement and to make appropriate changes where required, and to find new methods to fill gaps. In this presentation, the 'state of the art' in measurement of total dietary fibre and dietary fibre components will be described and discussed, together with suggestions for future research.
Hide AbstractMeasurement of resistant starch by enzymatic digestion in starch and selected plant materials: Collaborative study.
McCleary, B. V., McNally, M. & Rossiter, P. (2002). Journal of AOAC International, 85(5), 1103-1111.
Interlaboratory performance statistics was determined for a method developed to measure the resistant starch (RS) content of selected plant food products and a range of commercial starch samples. Food materials examined contained RS (cooked kidney beans, green banana, and corn flakes) and commercial starches, most of which naturally contain, or were processed to yield, elevated RS levels. The method evaluated was optimized to yield RS values in agreement with those reported for in vivo studies. Thirty-seven laboratories tested 8 pairs of blind duplicate starch or plant material samples with RS values between 0.6 (regular maize starch) and 64% (fresh weight basis). For matrixes excluding regular maize starch, repeatability relative standard deviation (RSDr) values ranged from 1.97 to 4.2%, and reproducibility relative standard deviation (RSDR) values ranged from 4.58 to 10.9%. The range of applicability of the test is 2-64% RS. The method is not suitable for products with <1% RS (e.g., regular maize starch; 0.6% RS). For such products, RSDr and RSDR values are unacceptably high.
Hide AbstractMeasurement of resistant starch.
McCleary, B. V. & Monaghan, D. A. (2002). Journal of AOAC International, 85(3), 665-675.
A robust and reliable method was developed to measure resistant starch (RS), i.e., starch that enters the large intestine. In vivo conditions were reflected as much as possible while a user-friendly format was maintained. Parameters investigated included α-amylase concentration, pH of incubation, maltose inhibition of α-amylase, the need for amyloglucosidase inclusion, the effect of shaking and stirring on determined values, and problems in recovering and analyzing the RS-containing pellet. The RS values obtained were in good agreement with published in vivo data. An interlaboratory evaluation of the method has been completed (First Action Method 2002.02).
Hide AbstractMeasurement of α-amylase activity in white wheat flour, milled malt, and microbial enzyme preparations, using the ceralpha assay: Collaborative study.
McCleary, B. V., McNally, M., Monaghan, D. & Mugford, D. C. (2002). Journal of AOAC International, 85(5), 1096-1102.
This study was conducted to evaluate the method performance of a rapid procedure for the measurement of α-amylase activity in flours and microbial enzyme preparations. Samples were milled (if necessary) to pass a 0.5 mm sieve and then extracted with a buffer/salt solution, and the extracts were clarified and diluted. Aliquots of diluted extract (containing α-amylase) were incubated with substrate mixture under defined conditions of pH, temperature, and time. The substrate used was nonreducing end-blocked p-nitrophenyl maltoheptaoside (BPNPG7) in the presence of excess quantities of thermostable α-glucosidase. The blocking group in BPNPG7 prevents hydrolysis of this substrate by exo-acting enzymes such as amyloglucosidase, α-glucosidase, and β-amylase. When the substrate is cleaved by endo-acting α-amylase, the nitrophenyl oligosaccharide is immediately and completely hydrolyzed to p-nitrophenol and free glucose by the excess quantities of α-glucosidase present in the substrate mixture. The reaction is terminated, and the phenolate color developed by the addition of an alkaline solution is measured at 400 nm. Amylase activity is expressed in terms of Ceralpha units; 1 unit is defined as the amount of enzyme required to release 1 µmol p-nitrophenyl (in the presence of excess quantities of α-glucosidase) in 1 min at 40°C. In the present study, 15 laboratories analyzed 16 samples as blind duplicates. The analyzed samples were white wheat flour, white wheat flour to which fungal α-amylase had been added, milled malt, and fungal and bacterial enzyme preparations. Repeatability relative standard deviations ranged from 1.4 to 14.4%, and reproducibility relative standard deviations ranged from 5.0 to 16.7%.
Hide AbstractTwo issues in dietary fiber measurement.
McCleary, B. V. (2001). Cereal Foods World, 46, 164-165.
Enzyme activity and purity of these topics, the easiest to deal with is the importance of enzyme purity and activity. As a scientist actively involved in polysaccharide research over the past 25 years, I have come to appreciate the importance of enzyme purity and specificity in polysaccharide modification and measurement (7). These factors translate directly to dietary fiber (DF) methodology, because the major components of DF are carbohydrate polymers and oligomers. The committee report published in the March issue of Cereal FOODS WORLD refers only to the methodology for measuring enzyme purity and activity (8) that led up the AOAC method 985.29 (2). In this work enzyme purity was gauged by the lack of hydrolysis (i.e., complete recovery) of a particular DF component (e.g. β-glucan, larch galactan or citrus pectin). Enzyme activity was measured by the ability to completely hydrolyze representative starch and protein (namely wheat starch and casein). These requirements and restrictions on enzyme purity and activity were adequate at the time the method was initially developed and served as a useful working guide. However, it was recognized that there was a need for more stringent quality definitions and assay procedures for enzymes used in DF measurements.
Hide AbstractNew developments in the measurement of α-amylase, endo-protease, β-glucanase and β-xylanase.
McCleary, B. V. & Monaghan, D. (2000). “Proceedings of the Second European Symposium on Enzymes in Grain Processing”, (M. Tenkanen, Ed.), VTT Information Service, pp. 31-38.
Over the past 8 years, we have been actively involved in the development of simple and reliable assay procedures, for the measurement of enzymes of interest to the cereals and related industries. In some instances, different procedures have been developed for the measurement of the same enzyme activity (e.g. α-amylase) in a range of different materials (e.g. malt, cereal grains and fungal preparations). The reasons for different procedures may depend on several factors, such as the need for sensitivity, ease of use, robustness of the substrate mixture, or the possibility for automation. In this presentation, we will present information on our most up-to-date procedures for the measurement of α-amylase, endo-protease, β-glucanase and β-xylanase, with special reference to the use of particular assay formats in particular applications.
Hide AbstractMeasuring dietary fibre.
McCleary, B. V. (1999). The World of Ingredients, 50-53.
Interest in dietary fibre is undergoing a dramatic revival thanks in part to the introduction of new carbohydrates as dietary fibre components. Much emphasis is being placed on determining how much fibre is present in a food. Linking a particular amount of fibre to a specific health benefit is now an important area of research. Total Dietary Fibre. The term “dietary fibre” first appeared in 1953 and referred to hemicelluloses, celluloses and lignin (1). In 1974, Trowell (2) recommended this term as a replacement for the no longer acceptable term “crude fibre” Burkitt (3) has likened the interest in dietary fibre to the growth of a river from its first trickle to a mighty torrent. He observes that dietary fibre “was viewed as merely the less digestible constituent of food which exerts a laxative action by irritating the gut “thus acquiring the designation “roughage” a term which was later replaced by “crude fibre” and ultimately by “dietary fibre” Various definitions of dietary fibre have appeared over the years, partly due the various concepts used in deriving the term (i.e. origin of material, resistance to digestion, fermentation in the colon etc.), and partly to the difficulties associated with its measurement and labelling (4). The principle components of dietary fibre, as traditionally understood, are non-starch polysaccharides, which in plant fibre are principally hemicelluloses and celluloses, and the non-carbohydrate phenolic components, cutin, suberin and waxes with which they are associated in Nature.
Hide AbstractSheehan, H. & McCleary, B. V. (1988). Biotechnology Techniques, 2(4), 289-292.
A procedure for the measurement of fungal and bacterial α-amylase in crude culture filtrates and commercial enzyme preparations is described. The procedure employs end-blocked (non-reducing end) p-nitrophenyl maltoheptaoside in the presence of amyloglucosidase and α-glucosidase, and is absolutely specific for α-amylase. The assay procedure is simple, reliable and accurate.
Hide AbstractMeasurement of cereal α-Amylase: A new assay procedure.
McCleary, B. V. & Sheehan, H. (1987). Journal of Cereal Science, 6(3), 237-251.
A new procedure for the assay of cereal α-amylase has been developed. The substrate is a defined maltosaccharide with an α-linked nitrophenyl group at the reducing end of the chain, and a chemical blocking group at the non-reducing end. The substrate is completely resistant to attack by β-amylase, glucoamylase and α-glucosidase and thus forms the basis of a highly specific assay for α-amylase. The reaction mixture is composed of the substrate plus excess quantities of α-glucosidase and glucoamylase. Nitrophenyl-maltosaccharides released on action of α-amylase are instantaneously cleaved to glucose plus free p-nitrophenol by the glucoamylase and α-glucosidase, such that the rate of release of p-nitrophenol directly correlates with α-amylase activity. The assay procedure shows an excellent correlation with the Farrand, the Falling Number and the Phadebas α-amylase assay procedures.
Hide AbstractImpact of starter culture on biochemical properties of sourdough bread related to composition and macronutrient digestibility.
Kulathunga, J., Whitney, K. & Simsek, S. (2023). Food Bioscience, 53, 102640.
Due to the desire for nutritionally dense foods, there is interest in whole grain and sourdough bread products. This research study was carried out to evaluate the effect of different starter cultures and stone mill settings on the nutritional attributes of sourdough bread. Using a stone mill, a blend of three hard red spring wheat varieties was ground into whole-grain flour. Three different gap settings and two different rotation speed values were used to produce flour samples with six different treatments, and those six flour samples were used to make sourdough bread with two different starter cultures. The experimental design was a randomized complete block design with a factorial arrangement. Significant differences (p < 0.05) were observed in dietary fiber content, arabinoxylan content, total starch content, and protein digestibility between sourdough bread made with rye and wheat starter cultures. Higher protein digestibility values were observed for most of the sourdough samples with the rye starter culture compared to the wheat starter culture. In addition, the protein digestibility showed a strong negative correlation (r = −0.85) with the coarse particles and a positive correlation (r = 0.70) with the fine particle size fraction. These results demonstrated that selected starter cultures and the raw material with the desired particle size could improve sourdough bread's nutritional attributes and potential application in the baking industry.
Hide AbstractStudy on nutritional components evaluation and origin differences of Agrocybe cylindracea from different regions.
Chen, L., Chen, Y., Wang, J., Wang, Z., & Huang, X. (2023). eFood, 4(2), e81.
The aim of this study is to investigate the nutrients of Agrocybe cylindracea from different regions and discover the law of its nutritional value. In this study, 43 A. cylindracea samples were collected from 13 provinces in China. The differences in nutritional components of this A. cylindracea were explored. The main nutritional components, including protein, dietary fiber, amino acids, fat, ash, and moisture, were evaluated. Further factor and cluster analysis were applied to analyze these nutrients. The results showed A. cylindracea had high protein (14.09–27.24 g/100 g dw) and insoluble dietary fiber (25.27–34.17 g/100 g dw), but low soluble dietary fiber (0.49–3.19 g/100 g dw) and fat (1.69–3.45 g/100 g dw). Seventeen amino acids were detected from these mushrooms, of which glutamic (18.24–46.78 g/100 g dw) was the most abundant. Based on Eta analysis, factor analysis, and cluster analysis, it was found that the comprehensive nutritional value of A. cylindracea was related to the factors of both the geographical origin and maturity stage. In addition, geographical origin had a greater influence on the nutritional value of A. cylindracea than the maturity stage. This study will serve as a useful reference for the selection of the consumption and further processing directions of A. cylindracea.
Hide AbstractPotential Antitumor Effect of Functional Yogurts Formulated with Prebiotics from Cereals and a Consortium of Probiotic Bacteria.
Ciric, A., Radu, N., Zaharie, M. G. O., Neagu, G., Pirvu, L. C., Begea, M. & Stefaniu, A. (2023). Foods, 12(6), 1250.
Various types of functional yogurts were obtained from normalized milk (with normalized lipid content) and a standardized probiotic consortium of probiotic bacteria named ABY3. All the types of yogurts obtained contained prebiotics from black or red rice; malt of barley, rye, wheat; or wheat bran. The physico-chemical analyses of all the functionalized products obtained showed that all of them met the quality standard for yogurt products. However, the sensorial analyses showed that the products obtained from black and red rice were of very good quality. The biological analyses indicated that all the types of products contained live probiotic bacteria, but wheat bran and red rice could increase their numbers. Tests performed on tumor cell line Caco-2 with corresponding postbiotics revealed cytotoxicity greater than 30% after 48 h of exposure in the case of yogurts obtained from milk with 0.8% lipid content and red rice or blond malt of barley. In the case of yogurts derived from milk with 2.5% lipid content, only the variants that contained blond malt of rye or wheat became cytotoxic against the Caco-2 cell line.
Hide AbstractModulation of gut microbiota by rice starch enzymatically modified using amylosucrase from Deinococcus geothermalis.
Song, E. J., Lee, E. S., So, Y. S., Lee, C. Y., Nam, Y. D., Lee, B. H. & Seo, D. H. (2023). Food Science and Biotechnology, 32(4), 565-575.
Amylosucrase can increase the amount of resistant starch (RS) in starch by transferring glucose from sucrose to amylopectin. Here, rice starch was modified using amylosucrase from Deinococcus geothermalis (DgAS). DgAS-modified rice starch (DMRS) increased the side-chain length of amylopectin and appeared in the form of B-type crystals. In vitro digestion analyses revealed that DMRS had a higher RS contents and lower digestion rate than native rice starch. When high-fat diet (HFD)-induced C57BL/6 mice were orally administered DMRS, body weight and white fat tissues of DMRS-fed HFD mice were not significantly different. However, serum leptin and glucose levels were significantly decreased and serum glucagon like peptide-1was increased in these mice. The cecal microbiome in DMRS-fed HFD mice was identified to investigate the role of DMRS in gut microbiota regulation. DMRS supplementation increased the relative abundance of Bacteroides, Faecalibaculum, and Ruminococcus in mouse gut microbiota.
Hide Abstract| Symbol : | GHS08 |
| Signal Word : | Danger |
| Hazard Statements : | H334 |
| Precautionary Statements : | P261, P284, P304+P340, P342+P311, P501 |








